 Written by: Genya Gorshtein, MSc
Written by: Genya Gorshtein, MSc
Published: October 16, 2024
Introduction
Understanding antibody-antigen interactions is critical for therapeutic and diagnostic development, where precise targeting of antibodies can lead to more effective treatments and reliable diagnostic tools.
This discussion will explore different epitope mapping methods, highlighting their advantages and limitations for mapping antibody-antigen interactions.
Epitope Mapping Methods
There are several epitope mapping techniques, including HDX-MS, XL-MS, Cryo-EM, X-ray crystallography, and fast photochemical oxidation of proteins (FPOP-MS), each offering distinct strengths and limitations. The principles, key advantages, and limitations of each method are summarized in Table 1.
For a comparison on HDX-MS and alanine scanning mutagenesis, visit our other article Epitope Mapping: Alanine Scanning Mutagenesis vs. HDX-MS.
Table 1. Summary of epitope mapping methods, key advantages, and limitations.
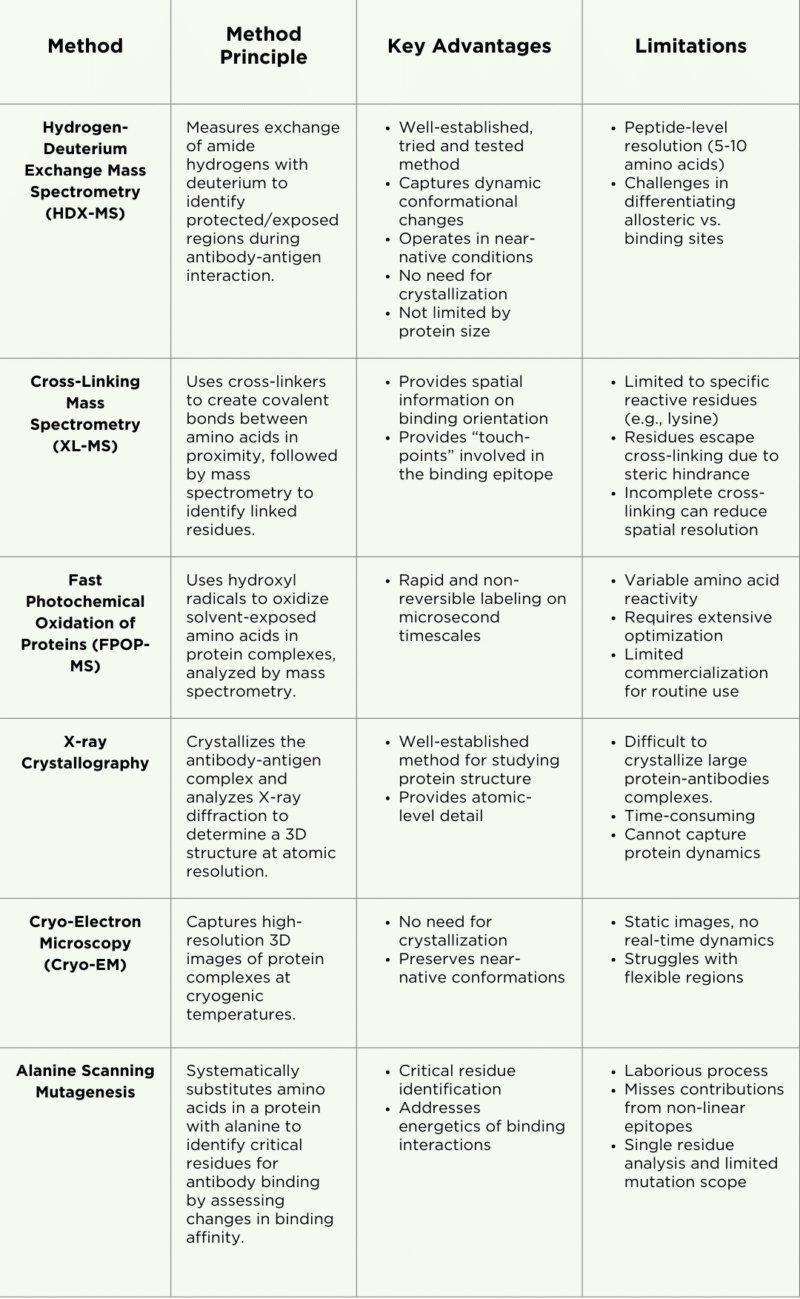
Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS)
HDX-MS is a mass spectrometry-based technique used to analyze antibody epitopes, protein structures, protein-protein interactions, and dynamic conformational changes. It leverages liquid chromatography coupled with mass spectrometry (LC-MS) to measure the exchange of amide hydrogen atoms with deuterium (D2O) along the protein backbone (Figure 1).
These exchanges offer insights into regions of the protein that are protected (less exchange) and exposed (more exchange). By comparing deuteration levels between an antigen’s antibody-bound and free states, areas shielded by antibody binding can be identified. For instance, regions of the antigen that interact with the antibody show reduced deuterium uptake. This data helps reveal conformational changes and antigen dynamics upon antibody binding, enhancing our understanding of the interaction.
Strengths and Applications
HDX-MS is a versatile method widely applied in epitope mapping and in the study of protein allostery, complex protein conformations, protein aggregation, and intrinsically disordered proteins.
HDX-MS captures highly accurate structures that closely align with those obtained from X-ray crystallography. A significant advantage of HDX-MS is its ability to analyze proteins in near-native conditions, eliminating the need for the challenging and often impractical protein crystallization required by traditional methods like X-ray crystallography.
This technique is particularly effective at capturing dynamic interactions, such as allosteric sites and conformational changes, providing valuable insights into antibody-antigen interactions. Additionally, HDX-MS is not limited by the size of the protein or protein complex, further enhancing its utility in epitope mapping studies.
Limitations
While HDX-MS has some major advantages for uses in epitope mapping, this method comes with some limitations:
- Differentiating allosteric sites from epitope binding sites: The dynamic nature of HDX-MS can make it challenging to distinguish between allosteric sites and epitope binding sites, as both may exhibit changes in deuterium uptake and exchange. Care must be taken to understand the kinetics of the interaction and gather a time series of HDX data accordingly.
- Peptide resolution: HDX-MS provides information on binding epitopes at a peptide resolution of approximately 5-10 amino acids, which is less detailed compared to the atomic-level resolution offered by X-ray crystallography. However, the resolution can be improved by analyzing the overlapping peptides in the data. Enzyme selection and efficiency are critical.
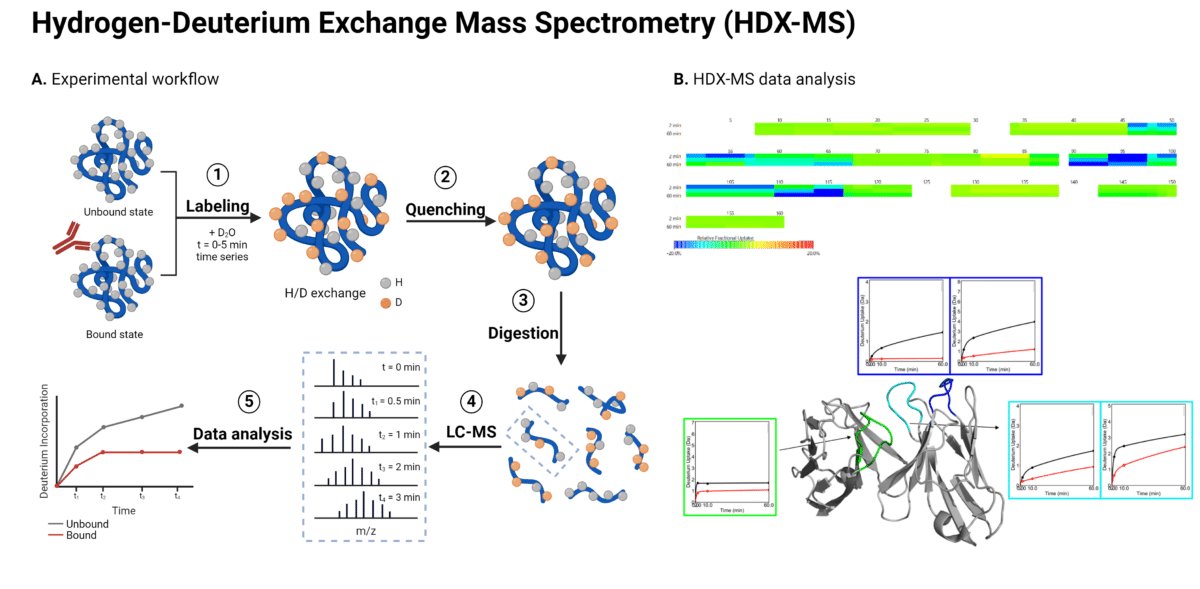
Figure 1. Workflow (A) and data analysis (B) for HDX-MS epitope mapping of antibody-antigen interactions. In the HDX-MS heat map depicting changes in deuterium uptake, green represents regions showing no change in deuterium uptake, cyan represents regions showing a slight decrease in deuterium uptake, and blue represents regions showing a strong decrease in deuterium uptake.
Cross-Linking Mass Spectrometry
Cross-linking mass spectrometry (XL-MS) involves the use of a chemical reagent known as a cross-linker, which reacts with two functional groups in a protein or protein complex (Figure 2). The cross-linker has a specific length and forms covalent bonds with particular amino acid side chains, enabling the identification of the linked amino acids through mass spectrometry. The positions of the cross-linked amino acids and the length of the cross-linker create distance constraints that help to refine the 3D modeled structure of the protein or the protein complex.
By incorporating these constraints into computational models, the protein’s structure can be derived, revealing the interaction interfaces.
Strengths and Applications
By chemically cross-linking specific amino acid side chains in proximity, XL-MS reveals how proteins are positioned relative to each other. While this spatial information can benefit molecular modeling and docking experiments, it is generally insufficient data for epitope mapping applications.
Limitations
A limitation of XL-MS is that it underestimates the epitope region, identifying only individual amino acid ‘touch-points’ rather than the continuous binding epitopes. Two critical limitations of XL-MS contribute to this:
- Limited cross-linker reactivity: Cross-linkers, such as disuccinimidyl suberate (DSS), primarily react with lysine and can only identify cross-links and interactions within their linker length (10–12 Å for DSS, for example). For XL-MS detection, the binding region must contain at least one lysine residue in close proximity to the binding interaction.
- Steric hindrance: Residues tightly packed in the binding interface may escape detection due to steric hindrance, reducing the precision of XL-MS in crowded or shielded regions.
Another limitation of XL-MS is incomplete cross-linking, which can lead to incomplete or variable spatial resolution that does not always align with X-ray crystal structures. For instance, a study published in 2024 investigated the use of XL-MS for epitope mapping of antigen-antibody complexes across three different antibody-antigen pairs. The authors reported variable agreement with crystal structures, with only 75%, 100%, or 42% of cross-links mapping successfully to the crystal structure.
Fast Photochemical Oxidation of Protein (FPOP-MS)
Fast photochemical oxidation of proteins (FPOP) is a mass spectrometry-based approach for studying protein structures and interactions (Figure 2). FPOP-MS maps epitope using hydroxyl radical protein footprinting (HRPF) to selectively oxidize solvent-exposed amino acids in an antibody-antigen complex. A UV laser generates these radicals from hydrogen peroxide, which reacts with accessible residues, while buried or tightly bound regions remain unmodified. After quenching the reaction, the protein is digested into peptides for mass spectrometry analysis. The oxidation patterns reveal exposed residues during binding, offering insights into the antibody-antigen interaction.
Strengths and Applications
While similar in concept to deuterium labeling in HDX-MS techniques, HRPF labeling is irreversible. This allows more flexibility after sample labeling for downstream processing, such as using longer chromatography gradients or enrichment strategies. FPOP labeling occurs on a much faster timescale than deuterium exchange, at the microsecond level – faster than proteins can fold, making it ideal for studying fast protein dynamics. This speed isn’t always necessary for epitope mapping between antibodies and antigens.
Limitations
FPOP-MS is a complex and expensive method for epitope mapping, presenting several challenges:
- Variable reactivity: Different amino acids show varying reactivity to hydroxyl radicals, with cysteine being the most reactive, followed by methionine. This can lead to residue modifications regardless of solvent accessibility, complicating data interpretation. Additionally, nearby secondary structures and neighboring residues can influence reactivity, adding another layer of complexity.
- High optimization needs: FPOP-MS requires significant optimization, accounting for variable reactivity and buffer compatibility with hydrogen radicals. This demands substantial time and resources before generating usable data.
- Limited commercialization: FPOP-MS is not yet widely commercialized. Unlike HDX-MS, which benefits from advanced analytical software and automated systems, FPOP-MS lacks these tools, limiting its ease of use and broader adoption for routine epitope mapping.
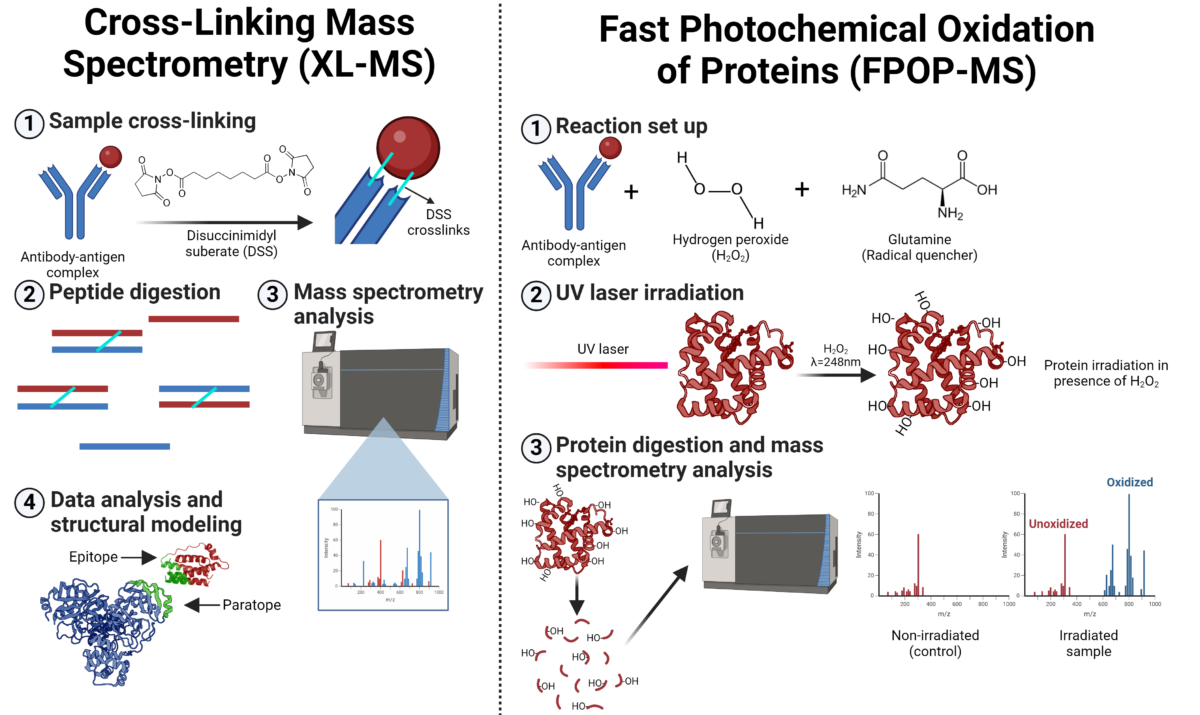
Figure 2. Workflow for cross-linking mass spectrometry (XL-MS) and fast photochemical oxidation of proteins mass spectrometry (FPOP-MS) for epitope mapping.
X-ray Crystallography
X-ray crystallography is a widely used technique for determining the 3D structures of biological molecules. By crystallizing the protein/complex and analyzing the diffraction pattern created when X-rays pass through it, a highly detailed 3D structure is generated. This structure reveals the contact points between the antibody and antigen at atomic resolution, enabling precise mapping of the epitope and the paratope.
X-ray crystallography provides valuable insights into the shape, orientation, and specific interactions involved in the binding, such as hydrogen bonds, van der Waals forces, and sometimes water-mediated interactions.
Strengths and Applications
Historically, X-ray crystallography has been regarded as the gold standard for structural protein analysis and is widely used across various disciplines and applications. Its primary strength lies in its ability to resolve atomic-level details, enabling researchers to visualize the precise arrangement of amino acids at the binding interface.
Limitations
X-ray crystallography presents significant limitations for epitope mapping analysis. Due to their large size, glycosylation, and multi-domain structure, antibodies are notoriously difficult to crystallize. This complexity makes studying antibody-antigen interactions via X-ray crystallography both challenging and time-consuming. As a result, smaller antibody fragments, such as single-chain antibodies (scFv), are often more suitable for crystallographic studies.
Additionally, X-ray crystallography does not capture the dynamic nature of these interactions, particularly the conformational changes that may occur during binding.
Cryo-Electron Microscopy
Cryo-Electron Microscopy (Cryo-EM) is a technique that uses an electron microscope to capture images of biological samples at cryogenic temperatures (Figure 3).
Single-particle cryo-EM generates a 3D view of macromolecules and complexes by averaging multiple particle images obtained through transmission electron microscopy at ultra-low temperatures. Thousands of particles, frozen in random orientations within vitreous ice, are sorted, classified, aligned, and averaged to reconstruct a detailed 3D model, which is then refined for high-resolution structural analysis.
Strengths and Applications
Unlike X-ray crystallography, cryo-EM allows for the observation of proteins in their near-native state without the need for crystallization. Samples are rapidly frozen in a thin layer of vitreous ice, preserving their native conformations and enabling researchers to study proteins under conditions that closely mimic their physiological environment.
Cryo-EM is particularly effective for visualizing large protein complexes, and advancements in technology are broadening its capabilities for analyzing smaller protein complexes as well. Additionally, cryo-EM is compatible with other structural techniques and is often used in conjunction with methods like HDX-MS to enhance the understanding of epitope mapping and provide complementary structural information.
Limitations
While cryo-EM excels at generating high-resolution structural information, especially for larger complexes, it provides a relatively static image of the protein complex. This means it doesn’t capture the subtle, real-time conformational changes that can occur during antibody-antigen binding.
Cryo-EM can struggle with resolving flexible or disordered regions, often resulting in low-resolution or missing density in final models. By combining cryo-EM with HDX-MS, researchers can obtain a more comprehensive understanding of protein dynamics and interactions.
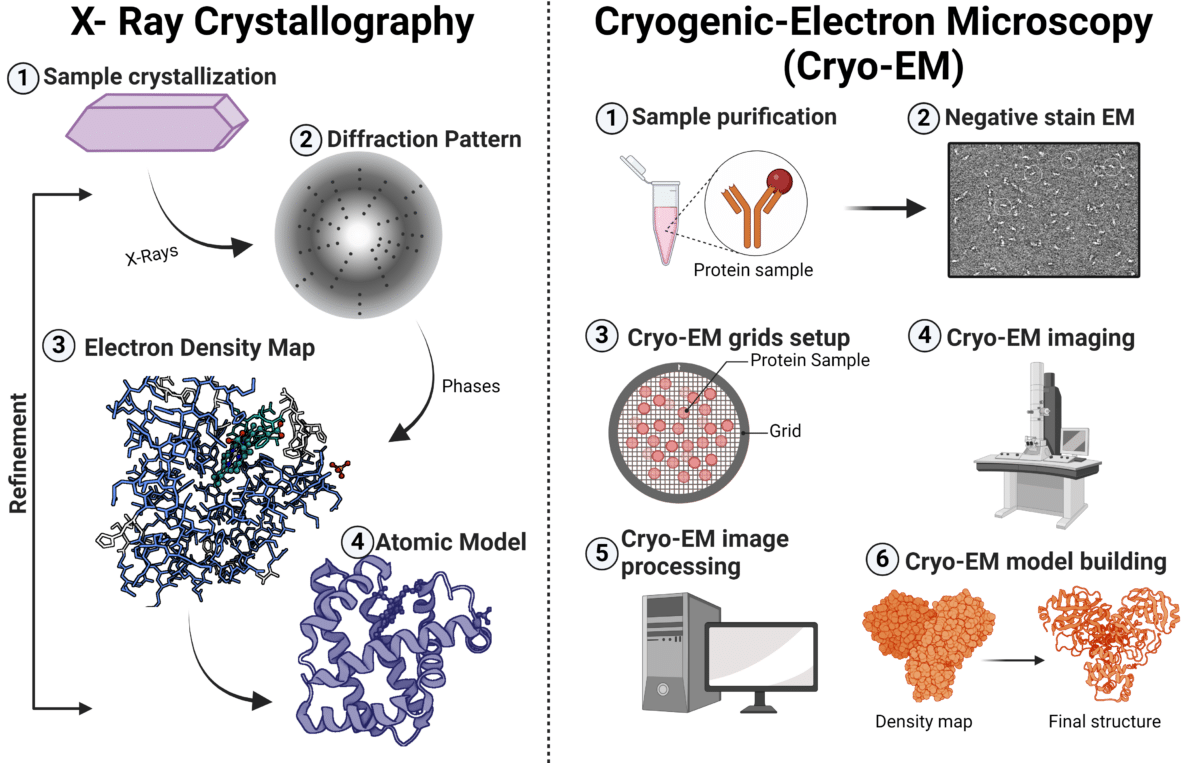
Figure 3. Workflow for X-ray crystallography and cryogenic-electron microscopy (Cryo-EM) for epitope mapping.
Addressing the Limitations of HDX-MS for Epitope Mapping
There are two primary limitations associated with using HDX-MS for epitope mapping of antibody-antigen interactions: differentiating allosteric sites from epitope binding sites and the peptide-level resolution.
While these limitations are noteworthy, they do not significantly affect the method’s ability to deliver accurate and reliable results for binding epitopes between antibodies and antigens.
Allosteric Sites vs. Epitope Binding
When studying antibody-antigen interactions, relying solely on static methods can be likened to looking at a picture of a racehorse—you can appreciate its form, but you can’t gauge its speed. While the dynamic nature of HDX-MS can present challenges, it is also a significant advantage.
Differentiating conformational changes from antibody binding in HDX-MS can be challenging, but expert evaluation of the data can provide valid conclusions on the binding epitope. A more significant change in deuterium uptake at short time points often suggests an epitope site, while smaller changes may indicate an allosteric or proximity site to an epitope site. Analyzing the kinetics of these changes also provides valuable information regarding whether a region is part of an allosteric site or directly involved in epitope binding.
To enhance the accuracy of these predictions, computational docking models, such as Rosetta, can be employed to confirm binding interactions and structural predictions, aiding in the differentiation of these complex dynamics.
Peptide-Level Resolution
As previously mentioned, HDX-MS provides epitope information at a peptide-level resolution, which can be enhanced by optimizing experiments to improve peptide coverage. This includes refining enzyme efficiency, adjusting conditions like pH and temperature, and employing multiple proteolytic enzymes and chromatography columns to achieve more comprehensive coverage.
What does resolution mean, and what level is needed?
The required resolution, whether peptide-level or atomic-level, depends on the specific application and end-point goals. For most antibody engineering and functional characterization purposes, peptide-level resolution from HDX-MS is sufficient. Since surrounding amino acids around the contact residues can contribute to the overall dynamics of antibody-antigen interactions, peptide-level resolution delivers comprehensive functional binding information. This insight is critical for guiding sequence and structural modifications during antibody engineering stages, ensuring effective design and optimization.
From an intellectual property (IP) perspective, having broad functional epitope claims can strengthen antibody patents. While claims based on specific residues offer precision, they can be vulnerable to design-arounds or minor modifications by competitors, potentially weakening patent protection. In contrast, broader epitope claims provides strong, more comprehensive protection by covering a wider range of amino acid sequences and conformational states that retain binding functionality.
HDX-MS as a Stand Alone Method for Epitope Mapping
HDX-MS has increasingly become the preferred method for epitope mapping, as evidenced by publication trends from PubMed indicating its rise over other techniques, such as X-ray crystallography (Figure 4). In contrast, as of 2024, there are relatively few studies utilizing cross-linking mass spectrometry (XL-MS) for epitope mapping of antigen-antibody complexes.
The widespread adoption of HDX-MS is further supported by advancements in mass spectrometry instrumentation, ultra-performance liquid chromatography (UPLC), automated sample handling, and sophisticated analysis software, making it an extremely dependable method.
For comprehensive epitope mapping, employing a complementary strategy that combines HDX-MS with a secondary method, is ideal; however, HDX-MS can effectively function as a standalone method for this purpose.
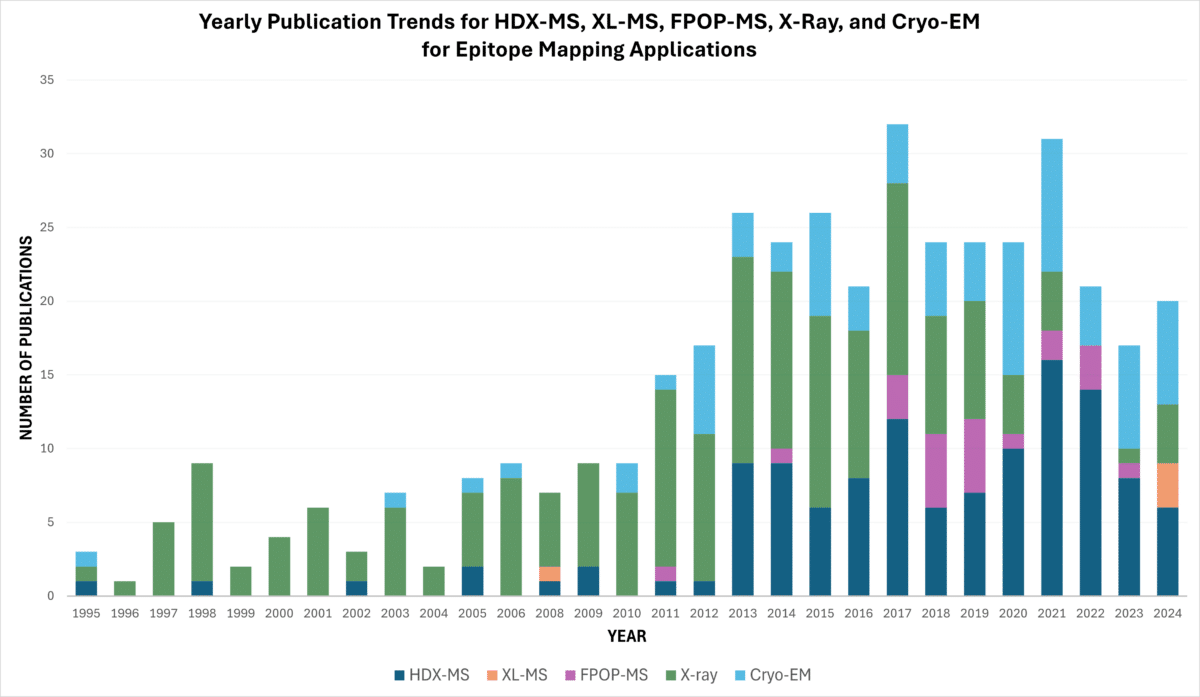
Figure 4. Yearly PubMed publication trends for HDX-MS, XL-MS, FPOP-MS, X-ray, and Cryo-EM epitope mapping applications as of October 2024, accessed from PubMed.
Epitope Mapping with HDX-MS at Rapid Novor
Rapid Novor offers an HDX-MS epitope mapping service that can accurately identify the binding site of an antibody to its corresponding antigen with high confidence and resolution. Utilizing the state-of-the-art Waters Cyclic IMS-MS and automated sample handling robotics, we achieve high throughput and peptide resolution of epitopes down to 1-5 amino acids. Our team includes PhD experts with over 7 years of experience in HDX-MS, working with a variety of analytes.
HDX-MS can also be integrated with AI in silico modeling, significantly increasing throughput and enhancing epitope validation.
Contact our scientists to learn more about epitope mapping using HDX-MS.

Talk to Our Scientists.
We Have Sequenced 10,000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and developed the first recombinant polyclonal antibody diagnostics.
Talk to Our Scientists.
We Have Sequenced 9000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables timely and reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and ran the first recombinant polyclonal antibody diagnostics