 Written by María Gerpe, PhD
Written by María Gerpe, PhD
August 17, 2021
Introduction
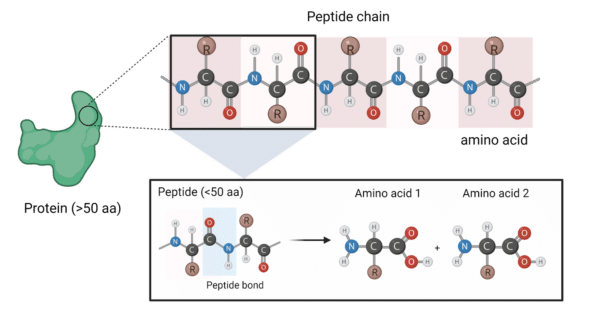
Amino acids (aa)—the building blocks of proteins—are simple molecules characterized by a variable R group flanked either side by an amino group and a carboxyl group. With around 20 different commonly found amino acids, each one can bond with another to produce chains that can be classified as peptides (typically below 50 aa) and proteins (sequences above 50 aa)—molecules ubiquitous to every known organism1.
Once the amino acid sequence of a protein (i.e., its primary structure) is established, it is possible to determine how the protein will fold and interact with other proteins. Information attained through protein sequencing is invaluable to the study of molecular biology and understanding of the basis of diseases. Two main methods are currently used to deduce the amino acid sequence of proteins: Edman degradation and mass spectrometry-based amino acid sequencing.
Edman Degradation
Developed by Swedish biochemist, Pehr Edman, over half a century ago2, Edman degradation involves the cyclical degradation of peptides that follows three main steps. Firstly, a reagent called phenyl isothiocyanate (PITC) is coupled to the α-amino group of a peptide or protein. The amino-terminal amino acid is then cleaved through cyclization and the resultant product is stabilized in aqueous acid to a phenylthiohydantion (PTH) amino acid. This process is repeated until the length of the amino acid has been degraded. The identity of each PTH-amino acid is determined using chromatographic or electrophoretic techniques, thereby enabling almost entire peptide sequences to be calculated.
While Edman degradation is not as popular as mass spectrometry-based sequencing methods, it can be useful in characterizing the N-terminus of a protein. Moreover, the method has since been miniaturized and coupled with gas-phase sequencers, expanding its power of application3.
However, Edman degradation still has some limitations. A common limitation of Edman degradation is that it is suited to sequencing short peptides (<30 to 50 residues) due to high error propensity. It is also low throughput (e.g., around 40 minutes per amino acid), and unable to sequence N-terminally inaccessible peptides (e.g., acetylated)2. This is why to capture the full sequence of a protein quickly, researchers often turn to mass spectrometry means.
Mass Spectrometry-based Protein Sequencing
Liquid chromatography-mass spectrometry (LC-MS) is a common method used to determine peptide sequences due to its ease of use and high-throughput workflows. Nowadays, liquid chromatography tandem mass spectrometry (LC-MS/MS is the preferred way to sequence proteins because of the additional fragmentations available, enabling higher accuracy and more complete information4. Both quantitative methods involve the enzymatic digestion of proteins into short peptide fragments prior to LC-MS or LC-MS/MS.
A mass spectrometer is used to ionize peptide molecules, separate them based on charge-to-mass ratios, and direct them towards an ion detector. The detector converts the flux of electrically-charged ions into a proportional electrical current processed into a mass spectrum by a data system. Using bioinformatic tools, the resulting sequence data can then be interpreted to resolve the original peptide sequences within a sample4.
Not only is protein mass spectrometry effective at identifying and fully sequencing a protein from the mass of its peptide fragment ions, it also lends itself to a range of applications. These include obtaining information on post-translational modifications and stoichiometry, characterizing protein structure, folding, function, and interactions, monitoring enzyme reactions, and quantifying proteins from a sample.
Summary
The importance of identifying protein sequences is well established. While Edman degradation is still used, mass spectrometry-based protein sequencing far exceeds the benefits from Edman sequencing. Contact a member of the Rapid Novor team today if you would like any more information.
References
- Essentials of Cell Biology, by Clare O’Connor et al. | The Online Books Page. https://onlinebooks.library.upenn.edu/webbin/book//lookupid?key=olbp65192.
- Edman, P. A method for the determination of amino acid sequence in peptides. Arch Biochem 22, 475 (1949).
- Alfaro, J. A. et al. The emerging landscape of single-molecule protein sequencing technologies. Nat Methods 18, 604–617 (2021).
- Hughes, C., Ma, B. & Lajoie, G. A. De novo sequencing methods in proteomics. Methods Mol Biol 604, 105–121 (2010).

Talk to Our Scientists.
We Have Sequenced 10,000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and developed the first recombinant polyclonal antibody diagnostics.
Talk to Our Scientists.
We Have Sequenced 9000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables timely and reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and ran the first recombinant polyclonal antibody diagnostics